
GtoPdb is requesting financial support from commercial users. Please see our sustainability page for more information.
Lanosterol biosynthesis pathway: Introduction

The lanosterol pathway is represented by Figure 1.
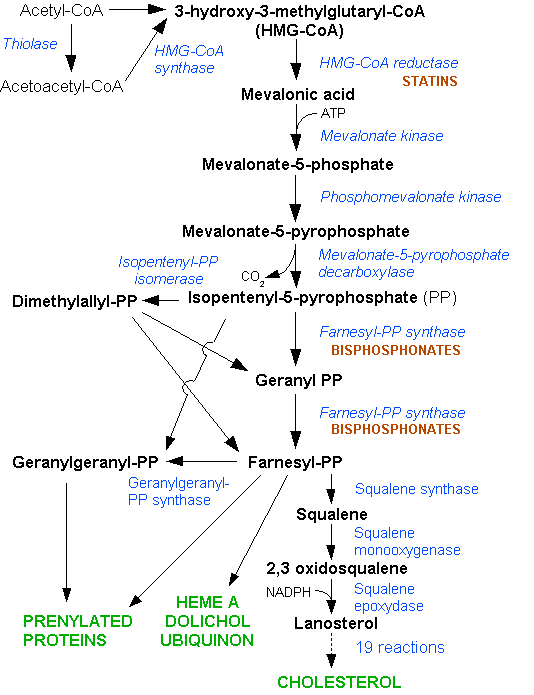
Figure 1: The lanosterol biosynthesis pathway Dodo, 2006 (Ref: http://en.wikipedia.org/wiki/File:HMG-CoA_reductase_pathway.png)
The lanosterol pathway is present in all higher eukaryote species, archeabacteria and eubacteria [27]. The pathway intermediates are the precursors for steroid hormones, polyisoprenoids and other molecules integral for cellular metabolism [14]. Apicomplexan protozoa, plants and gram negative bacterial species synthesise isoprenoids via the non-mevalonate or methylerythritol phosphate (MEP) pathway [27,35], with common intermediate metabolites isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate synthesised by different enzymes than in mammalian and other eukaryotic species.
The enzymes of the lanosterol pathway are common to all mammalian species and are well characterised in humans and rodents. The review article by Miziorki, 2011 outlines the active site residues identified for each of the mevalonate pathway enzymes [27]. Recombinant forms of the human enzymes, expressed in E. coli and other bacterial species are frequently used in assays. A significant number of compounds have been assayed with human, rat and mice enzymes to establish their role as potential enzyme inhibitors.
Physiological roles of the pathway
The cholesterol biosynthesis pathway has several important physiological functions. Its end metabolite cholesterol is the precursor of numerous physiologically important molecules including oxysterols and bile acids, and forms the basis of steroid hormone synthesis.
The lanosterol segment of the cholesterol pathway comprises the rate-limiting and committed steps of cholesterol biosynthesis, the reactions catalysed by hydroxymethylglutaryl-CoA reductase and squalene synthase respectively. This segment of the pathway also incorporates a side branch leading to protein prenylation and farnesylation, essential processes for cell growth and the formation of non-sterol compounds. The classes and functions of these non-sterol products of the cholesterol synthesis pathway are outlined by Houten et al., 2003 [23], and Goldstein and Brown, 1990 [17]. The products of the cholesterol biosynthesis pathway and its side branches are outlined in review articles by Waterman [39], and Charlton-Menys and Durrington [8]. The numerous end-products and intermediates described reveal the complexity of the overall pathway and its various segments, and reveals varying nomenclature for both the cholesterol biosynthesis pathway as a whole and the lanosterol segment in particular. The term ‘mevalonate pathway’ is used to describe the series of enzymes from hydroxymethylglutaryl-CoA synthase through to diphophosphomevalonate decarboxylase catalysing the formation and metabolism of intermediate metabolite mevalonate. The segment of the cholesterol synthesis pathway prior to lanosterol synthesis is commonly referred to as the isoprenoid synthesis pathway. The structure and function of isoprenoids, also known as terpenoids, are outlined in reviews by Miziorko, 2011 [27], and Waterman, 2006 [39].
Cholesterol biosynthesis principally occurs in hepatic cells, with high rates of synthesis also present in intestinal cells, and rapidly proliferating cells [12,31].
Pathophysiological roles
The cholesterol biosynthesis pathway is of significant biomedical interest owing to its involvement in multiple disease processes. Excessive cholesterol synthesis can lead to hypercholesterolemia, one of a cluster of pathological phenomena implicated in cardiovascular disease and the development of metabolic syndrome [40]. Treatments seeking to prevent hypercholesterolemia limit endogenous cholesterol synthesis involve reversible inhibition of the enzyme catalysing the rate-limiting step in the pathway; HMG-CoA reductase [21].
There is increasing research interest in the role cholesterol biosynthesis plays in bacterial reproduction in species displaying enzyme homology with mammals, with studies exploring how enzyme inhibition or mutation influences the passage of metabolites through the pathway [27]. These include squalene synthase inhibition in Trypanosoma cruzi [33] and mevalonate kinase inhibition in Staphylococcus aureus [36]. Inhibition of the non-mevalonate pathway has been explored as a therapeutic strategy against diseases such as tuberculosis [29].
Knockout studies in mice have demonstrated the phenotypic consequences of loss of function of the cholesterogenic enzymes, which include embryonic lethality [22].
Subcellular localisation
There are conflicting views in the literature over the subcellular localisation of the lanosterol segment of the cholesterol biosynthesis pathway. HMG-CoA reductase is localised to the endoplasmic reticulum, while each of the other enzymes are believed to have a cytosolic location. However, several studies support a peroxisomal location for some of these enzymes. Peroxisomal targeting sequence motifs have been identified in the carboxyl-terminal sequences, as described by Kovacs et al.,2007 [25]. These findings are supported by several other studies using a variety of experimental techniques [1-2,25]. However, the findings of these studies have been questioned by several papers as outlined by Waterman, 2006 [39], in which a cytosolic location for each of the lanosterol pathway enzymes with the exception of HMG- CoA reductase appears to be affirmed.
Enzyme nomenclature and reaction classification
The lanosterol pathway enzymes are listed according the IUBMB nomenclature guidance. Previous and unofficial names are also listed for each enzyme; these include abbreviated names often derived from the approved gene names. The majority of the synonyms listed are in common use in the literature. Each enzyme-catalysed reaction can is represented by an Enzyme Commission (EC) number. A guide to EC numbers and their usage can be found on the IUBMB Guide to Nomenclature (http://www.chem.qmul.ac.uk/iubmb/enzyme/). As the guide illustrates, each digit in the EC number represents an aspect of the enzyme’s action. The majority of the enzymes in the pathway are classified as transferases, denoted by the ‘2’ at the start of the E.C. numbers describing the reactions they catalyse. The EC numbers annotated next to the reaction schemes are hyperlinked to a detailed explanation of each enzyme’s EC number on the KEGG BRITE hierarchy page. Several of the reactions are reversible, denoted by an equals sign. It is important to note EC numbers are used as identifiers for the reactions of the enzymes themselves. This is particularly significant in the case of the enzymes geranylgeranyl diphosphate synthase and farnesyl diphosphate synthase, which can both catalyse more than one pathway reaction, with some overlap in function, outlined below. These examples of multiple enzyme function are denoted by assigning more than one EC number to each enzyme, illustrating how EC numbers cannot serve as unique identifiers for enzymes.
Substrates stereochemistry
The majority of the endogenous substrates have defined stereochemistry; in these cases the naturally-found isomer is specified in the reaction equation. Examples include (S)-3-hydroxy-3-methylglutaryl-CoA and (R)-mevalonate. The stereochemistry of lanosterol pathway metabolites is described by Dickschat, 2011 [11].
Enzyme inhibition
In addition to data on various synthetic organic compounds tested in assays for their potential as inhibitors of the lanosterol pathway enzymes, the inhibitors listed in this database include endogenous ligands demonstrated to act as feedback inhibitors in the pathway. These ligands are indicated by the endogenous ligand symbol and are annotated as feedback inhibitors. The examples of feedback inhibition given of the database illustrate how intermediate metabolites can exert upstream effects in the pathway.
HMG-CoA reductase, is the target if the statin group of drugs, the sole clinical strategy for cholesterol synthesis inhibition. However, there is interest in developing alternative strategies for cholesterol biosynthesis inhibition, including the potential for a multi-target inhibition strategy. In exploring strategies for pathway inhibition it is imperative to understand the downstream effects of inhibition, particularly given the interaction of the lanosterol pathway with other metabolic pathways via its side branches.
Acetyl-CoA acetyltransferase 1 and 2
Acetyl-CoA acetyltransferase catalyses the conversion of two molecules of acetyl-CoA into acetoacetyl CoA and coenzyme A. A mutation in the mitochondrial form of the enzyme, acetyl-CoA acetyltransferase 1 is associated with metabolic disorder alpha-methylacetoacetic aciduria [37], while deficiency of the cytosolic form, acetyl-CoA acetyltransferase 2, is implicated in impairment of motor development [18]. The two forms of the enzymes are encoded by separate genes, ACAT1 and ACAT2.
Hydroxymethylglutaryl-CoA synthase 1 and 2 (HMG-CoA synthase 1 and 2)
The first enzyme in the lanosterol pathway catalyses the synthesis of the metabolite (S)-3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) from condensation and acyl group transfer of substrates acetyl CoA and acetoacetyl CoA. This reaction is physiologically irreversible [27]. HMG-CoA synthase 1 is localised to the cytoplasm. A mitochondrial isoform of the enzyme, HMG-CoA synthase 2, arising from a separate gene also exists [27]. All data in the tables refers to the cytoplasmic isoform, HMG-CoA synthase 1. Unlike the other pathway enzymes, there is little available experimental data for potential inhibitory compounds of HMG-CoA synthase.
Hydroxymethylglutaryl-CoA reductase (HMG-CoA reductase)
The second enzyme of the lanosterol pathway catalyses the reduction of HMG-CoA to mevalonate, the intermediate metabolite from which the pathway derives one if its names. This is a two-stage reaction, initially forming the unstable metabolite intermediates mevaldyl-CoA and mevaldehyde before further reduction to mevalonate [27].
HMG-CoA reductase is a transmembrane glycoprotein consisting of eight transmembrane domains, spanning the endoplasmic reticulum membrane [9,24,32]. Target sequences for the endoplasmic reticulum have been identified to be associated with the enzyme [30]. The active site of the enzyme is located in the cytosol, with the carbohydrate components located in the lumen of the endoplasmic reticulum [24].
As the target for statins, HMG-CoA reductase is the only current therapeutic target in the pathway, in a single-enzyme inhibition strategy. Although proven successful treatments for hypercholesterolaemia, statin use is dose-limited and side effect profiles vary between patients [5,7,26]. This may partly be explained by polymorphisms in the HMG-CoA reductase gene which have been demonstrated to alter the pharmacodynamic profile of statins [26]. There is therefore an established clinical need to consider alternative cholesterol synthesis inhibition strategies.
In addition to their clinical use, statins have been widely used as experimental agents to increase our knowledge of the pathway enzymes and their regulation. Binding studies with statin drugs have enhanced understanding of the structure of HMG-CoA reductase and the conformation of the active site when bound to inhibitor [7]. The mechanism of action of the statins has been established across multiple species. Autoregulation of the pathway via feedback inhibition can be manipulated by statins. In normal physiological conditions, HMG-CoA reductase expression is decreased as a result of increased cellular concentrations of the latter intermediate pathway metabolites. Inhibition of HMG-CoA reductase by a statin leads to a deficit in downstream pathway metabolites and therefore reduced feedback inhibition. This subsequently leads to an increase in HMG-CoA reductase expression. However, the enzyme function is inhibited by the statin present [17]. Statin treatment can be used to induce expression of HMG-CoA synthase, HMG-CoA reductase, farnesyl diphosphate synthase and isopentenyl delta isomerase [28]. This has been demonstrated in hepatocyte and skeletal muscle tissues.
HMG-CoA reductase is subject to enzyme-mediated autoregulation and feedback inhibition. It is activated by the enzyme hydroxymethylglutaryl-CoA reductase (NADPH) kinase and deactivated by hydroxymethylglutaryl-CoA reductase (NADPH)-phosphatase [3], while feedback inhibition occurs by means of two mechanisms: sterol regulatory element binding proteins (SREBPs) and oxysterols [39]. The latter have been demonstrated to promote ubiquitination and degradation of HMG-CoA reductase [13]. There is interest in promoting enzyme degradation as an alternative strategy for pathway inhibition [4,9].
Mammalian HMG-CoA reductase activity has been show to follow a circadian cycle, with the highest activity levels seen in darkness in rodents [31].
Mevalonate Kinase
The phosphotransferase mevalonate kinase catalyses the conversion of mevalonate to 5-phosphomevalonate; an irreversible reaction and the first phosphorylation step in the pathway [14]. There are two known pathologies associated with deficiency of mevalonate kinase; Hyper-IgD syndrome and mevalonic aciduria. Both conditions are described as autoinflammatory and result from mutations in the MVK gene. Each of these pathologies are characterised by accumulation of the intermediate metabolite mevalonate, the endogenous substrate of mevalonate kinase. Mevalonate kinase is subject to feedback inhibition by farnesyl diphosphate and geranyl diphosphate, intermediate metabolites synthesised by enzymes geranylgeranyl diphosphate synthase and farnesyl diphosphate synthase [14]. Mevalonate kinase is localised to the cytosol [27]. The structure and binding site conformation of mevalonate kinase is described by Fu et al., 2008 [14], in a study that examines how activity levels vary between mutant and wild type forms of the enzyme.
Phosphomevalonate kinase
Phosphomevalonate kinase catalyses the second phosphorylation reaction in the pathway, the conversion of (R)-5-phosphomevalonate to (R)-5-diphosphomevalonate. This reaction is reversible [27].
Diphosphomevalonate decarboxylase
Diphosphomevalonate decarboxylase catalyses the conversion of (R)-5 diphosphomevalonate to isopentenyl diphosphate in an irreversible reaction. This reaction requires a divalent cation and ATP as a phosphate acceptor. Analogues of pathway metabolites such as fluorinated mevalonate 5-diphophate have been assayed as inhibitors of diphosphomevalonate decarboxylase, supporting their role in feedback inhibition in the pathway.
Isopentenyl diphosphate delta isomerase 1 (IDI1) and 2 (IDI2)
Isopentenyl diphosphate delta isomerase isoforms IDI1 and IDI2 are the products of separate genes, with mammalian IDI2 believed to have evolved as a result of IDI1 gene duplication, a theory supported by structural and sequence analysis [6]. Despite this duplication, IDI2 is not a naptogene, and is believed to have diverged rapidly from its isoform. The two isoforms display distinct tissue distribution patterns, with IDI2 expressed at high levels only in skeletal muscle and ID1 in other tissues including the brain and liver [6,10]. The results of one study demonstrate that cholesterol biosynthesis can be inhibited more readily in skeletal muscle than in other tissues [28].
Peroxisomal localisation is supported for both isoforms of the enzyme [10], although it has been suggested that the subcellular location of the two isoforms remains inconclusive [6]. While there is a high degree of sequence homology between the two isoforms of the enzyme and the reaction they catalyse is identical, the active site conformation is distinct in the two cases. A serine residue forming part of the active site in IDI1 is substituted by a cysteine residue in IDI2 [42].
Their role in the pathway is to catalyse the reversible inter-conversion of isopentenyl diphosphate and its nucleophilic isomer dimethylallyl diphosphate. The equilibrium of this reactions lies towards the product [44]. The two enzymes are regulated independently. The mechanism for IDI2 regulation is undetermined but believed to involve PPARα [10].
Geranylgeranyl diphosphate synthase and Farnesyl diphosphate synthase
Geranylgeranyl diphosphate synthase (GGPS1) is capable of catalysing more than one reaction in the pathway and therefore has more than one EC number assigned to it. This multiplicity of function is also observed in the case of farnesyl diphosphate synthase. Geranylgeranyl diphosphate synthase catalyses three reactions, while farnesyl diphosphate synthase catalyses two. The enzymes also display overlap in of function and are believed to have evolved from a common ancestor gene. This has been suggested as a result of studies in bacteria [34]. Mutagenesis experiments have also demonstrated inter-conversion of the two enzymes.
The two reactions catalysed by farnesyl diphosphate synthase are also catalysed by geranylgeranyl diphosphate synthase, and are represented by EC numbers 2.5.1.1 and 2.5.1.10. The third reaction catalysed by geranylgeranyl diphosphate synthase is represented by EC number 2.5.1.29. Geranylgeranyl diphosphate synthase activity forms geranylgeranyl diphosphate, a precursor in terpenoid biosynthesis, a side branch of the lanosterol pathway. Given the overlap in function with farnesyl diphosphate synthase and its role outside the lanosterol pathway, geranylgeranyl diphosphate synthase has not always been considered to truly be an enzyme of cholesterol biosynthesis.
The interconversion of dimethylallyl diphosphate and isopentenyl diphosphate into diphosphate and geranyl diphosphate is represented by EC number 2.5.5.1. Several databases assign this EC number to dimethylallyltranstransferase, an enzyme not present in mammals. This reaction has been demonstrated to be catalysed by both geranylgeranyl diphosphate synthase and farnesyl diphopshate synthase in mammalian species.
Geranylgeranyl diphosphate synthase and farnesyl diphosphate synthase are referred to as the branch point enzymes [23]. Farnesyl diphosphate is a precursor of numerous non-sterol products including including isoprenoids and farnesylated proteins [20]. From this stage in the pathway, metabolites either pass through the cholesterol biosynthesis pathway or are converted to non-sterol end products via a side branch. The products of the reactions catalysed by these two enzymes contribute to feedback inhibition of HMG-CoA reductase.
Inhibitors of farnesyl diphosphate synthase and geranylgeranyl diphosphate synthase include the bisphosphonate group of compounds; approved drugs used in the treatment of bone disorders [43]. Their mechanism of action is to prevent the passage of metabolites thought the protein prenylation side branch of the pathway through inhibition of farnesyl diphosphate synthase in osteoclasts [19]. They include zoledronate, a strong inhibitor of farnesyl diphopshate synthase and a weak inhibitor of geranylgeranyl diphosphate synthase [41].
Squalene synthase
The conversion of trans,trans-farnesyl diphosphate into squalene is known as the committed step in sterol biosynthesis. Squalene is then converted into lanosterol, forming the basis of sterol formation. A multitude of compounds have been assayed as potential squalene synthase inhibitors. Potent inhibitors include zaragozic acid, a natural product derived from fungi. Squalene synthase is localised to the ER membrane.
Squalene monooxygenase
Squalene monooxygenase catalyses the conversion of squalene to (S)-2,3-epoxysqualene. Thhis enzyme catalyses the first oxygenation step in sterol biosynthesis and like HMG-CoA reductase has been described as one of the rate-limiting enzymes of the pathway [16].
Lanosterol synthase
Lanosterol synthase catalyses the conversion of (S)-2,3-epoxysqualene to lanosterol. There is interest in the development of lanosterol synthase inhibitors as alternative therapies to statins in the treatment of hypercholesterolaemia, as lanosterol synthase occurs further downstream in the pathway therefore the effects of inhibiting this enzyme cause fewer effects upstream in the pathway, and its activity is more directly linked to the synthesis of cholesterol [8].
Following conversion of (S)-2,3-epoxysqualene into lanosterol, the intermediate metabolite passes through a series of further enzyme catalysed reactions before the formation of end product cholesterol. The post-lanosterol stage of the pathway is outlined in review articles by Gaylor 2002, [15]; Waterman, 2006 [39]; Waterman, 2002 [38]; Herman, 2003 [20]. The 2006 review by Waterman discusses the possibility of the stages of conversion from lanosterol to cholesterol occurring in a different sequence depending on the tissue in which the enzymes are expressed. Eight enzymes have been shown to be involved in the post-squalene stages of cholesterol synthesis and in some cases are capable to acting on more than one substrate. These reviews also discuss pathologies resulting from enzyme deficiencies in the post-squalene segment of the pathway. Fakheri and Javitt, 2011 describe the post-lanosterol section of cholesterol synthesis as having nineteen stages and discuss autoregulation of the pathway and its consequences [13].
References
1. Aboushadi N, Engfelt WH, Paton VG, Krisans SK. (1999) Role of peroxisomes in isoprenoid biosynthesis. J Histochem Cytochem, 47 (9): 1127-32. [PMID:10449533]
2. Bahmer FA, Feldmann U. (1995) Objective and reproducible assessment of irritants in vivo. A reappraisal of the IT50 in honour of Kligman and Wooding. Curr Probl Dermatol, 23: 288-95. [PMID:9035924]
3. Beg ZH, Stonik JA, Brewer HB. (1979) Characterization and regulation of reductase kinase, a protein kinase that modulates the enzymic activity of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. Proc Natl Acad Sci USA, 76 (9): 4375-9. [PMID:291971]
4. Berkhout TA, Simon HM, Patel DD, Bentzen C, Niesor E, Jackson B, Suckling KE. (1996) The novel cholesterol-lowering drug SR-12813 inhibits cholesterol synthesis via an increased degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem, 271 (24): 14376-82. [PMID:8662919]
5. Blanc M, Hsieh WY, Robertson KA, Watterson S, Shui G, Lacaze P, Khondoker M, Dickinson P, Sing G, Rodríguez-Martín S et al.. (2011) Host defense against viral infection involves interferon mediated down-regulation of sterol biosynthesis. PLoS Biol, 9 (3): e1000598. [PMID:21408089]
6. Breitling R, Laubner D, Clizbe D, Adamski J, Krisans SK. (2003) Isopentenyl-diphosphate isomerases in human and mouse: evolutionary analysis of a mammalian gene duplication. J Mol Evol, 57 (3): 282-91. [PMID:14629038]
7. Carbonell T, Freire E. (2005) Binding thermodynamics of statins to HMG-CoA reductase. Biochemistry, 44 (35): 11741-8. [PMID:16128575]
8. Charlton-Menys V, Durrington PN. (2007) Squalene synthase inhibitors : clinical pharmacology and cholesterol-lowering potential. Drugs, 67 (1): 11-6. [PMID:17209661]
9. Chun KT, Simoni RD. (1992) The role of the membrane domain in the regulated degradation of 3-hydroxy-3-methylglutaryl coenzyme A reductase. J Biol Chem, 267 (6): 4236-46. [PMID:1740463]
10. Clizbe DB, Owens ML, Masuda KR, Shackelford JE, Krisans SK. (2007) IDI2, a second isopentenyl diphosphate isomerase in mammals. J Biol Chem, 282 (9): 6668-76. [PMID:17202134]
11. Dickschat JS. (2011) Isoprenoids in three-dimensional space: the stereochemistry of terpene biosynthesis. Nat Prod Rep, 28 (12): 1917-36. [PMID:21979838]
12. Dietschy JM, Turley SD, Spady DK. (1993) Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res, 34 (10): 1637-59. [PMID:8245716]
13. Fakheri RJ, Javitt NB. (2011) Autoregulation of cholesterol synthesis: physiologic and pathophysiologic consequences. Steroids, 76 (3): 211-5. [PMID:20951718]
14. Fu Z, Voynova NE, Herdendorf TJ, Miziorko HM, Kim JJ. (2008) Biochemical and structural basis for feedback inhibition of mevalonate kinase and isoprenoid metabolism. Biochemistry, 47 (12): 3715-24. [PMID:18302342]
15. Gaylor JL. (2002) Membrane-bound enzymes of cholesterol synthesis from lanosterol. Biochem Biophys Res Commun, 292 (5): 1139-46. [PMID:11969204]
16. Gill S, Stevenson J, Kristiana I, Brown AJ. (2011) Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab, 13 (3): 260-73. [PMID:21356516]
17. Goldstein JL, Brown MS. (1990) Regulation of the mevalonate pathway. Nature, 343 (6257): 425-30. [PMID:1967820]
18. Groot CJ, Haan GL, Hulstaert CE, Hoomes FA. (1977) A patient with severe neurologic symptoms and acetoacetyl-CoA thiolase deficiency. Pediatr Res, 11 (10 Pt 2): 1112-6. [PMID:20597]
19. Guo RT, Cao R, Liang PH, Ko TP, Chang TH, Hudock MP, Jeng WY, Chen CK, Zhang Y, Song Y et al.. (2007) Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc Natl Acad Sci USA, 104 (24): 10022-7. [PMID:17535895]
20. Herman GE. (2003) Disorders of cholesterol biosynthesis: prototypic metabolic malformation syndromes. Hum Mol Genet, 12 Spec No 1: R75-88. [PMID:12668600]
21. Holdgate GA, Ward WH, McTaggart F. (2003) Molecular mechanism for inhibition of 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) reductase by rosuvastatin. Biochem Soc Trans, 31 (Pt 3): 528-31. [PMID:12773150]
22. Horvat S, McWhir J, Rozman D. (2011) Defects in cholesterol synthesis genes in mouse and in humans: lessons for drug development and safer treatments. Drug Metab Rev, 43 (1): 69-90. [PMID:21247357]
23. Houten SM, Frenkel J, Waterham HR. (2003) Isoprenoid biosynthesis in hereditary periodic fever syndromes and inflammation. Cell Mol Life Sci, 60 (6): 1118-34. [PMID:12861380]
24. Jingami H, Brown MS, Goldstein JL, Anderson RG, Luskey KL. (1987) Partial deletion of membrane-bound domain of 3-hydroxy-3-methylglutaryl coenzyme A reductase eliminates sterol-enhanced degradation and prevents formation of crystalloid endoplasmic reticulum. J Cell Biol, 104 (6): 1693-704. [PMID:3584246]
25. Kovacs WJ, Tape KN, Shackelford JE, Duan X, Kasumov T, Kelleher JK, Brunengraber H, Krisans SK. (2007) Localization of the pre-squalene segment of the isoprenoid biosynthetic pathway in mammalian peroxisomes. Histochem Cell Biol, 127 (3): 273-90. [PMID:17180682]
26. Maggo SD, Kennedy MA, Clark DW. (2011) Clinical implications of pharmacogenetic variation on the effects of statins. Drug Saf, 34 (1): 1-19. [PMID:21142270]
27. Miziorko HM. (2011) Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch Biochem Biophys, 505 (2): 131-43. [PMID:20932952]
28. Morikawa S, Murakami T, Yamazaki H, Izumi A, Saito Y, Hamakubo T, Kodama T. (2005) Analysis of the global RNA expression profiles of skeletal muscle cells treated with statins. J Atheroscler Thromb, 12 (3): 121-31. [PMID:16020911]
29. Obiol-Pardo C, Rubio-Martinez J, Imperial S. (2011) The methylerythritol phosphate (MEP) pathway for isoprenoid biosynthesis as a target for the development of new drugs against tuberculosis. Curr Med Chem, 18 (9): 1325-38. [PMID:21366531]
30. Olender EH, Simon RD. (1992) The intracellular targeting and membrane topology of 3-hydroxy-3-methylglutaryl-CoA reductase. J Biol Chem, 267 (6): 4223-35. [PMID:1740462]
31. Polo M, de Bravo MG, Carbone C. (1999) 3-Hydroxy-3-methylglutaryl coenzyme a reductase activity in liver of athymic mice with or without an implanted human carcinoma. Comp Biochem Physiol B, Biochem Mol Biol, 122 (4): 433-7. [PMID:10392455]
32. Roitelman J, Olender EH, Bar-Nun S, Dunn WA, Simoni RD. (1992) Immunological evidence for eight spans in the membrane domain of 3-hydroxy-3-methylglutaryl coenzyme A reductase: implications for enzyme degradation in the endoplasmic reticulum. J Cell Biol, 117 (5): 959-73. [PMID:1374417]
33. Sealey-Cardona M, Cammerer S, Jones S, Ruiz-Pérez LM, Brun R, Gilbert IH, Urbina JA, González-Pacanowska D. (2007) Kinetic characterization of squalene synthase from Trypanosoma cruzi: selective inhibition by quinuclidine derivatives. Antimicrob Agents Chemother, 51 (6): 2123-9. [PMID:17371809]
34. Sitthithaworn W, Kojima N, Viroonchatapan E, Suh DY, Iwanami N, Hayashi T, Noji M, Saito K, Niwa Y, Sankawa U. (2001) Geranylgeranyl diphosphate synthase from Scoparia dulcis and Croton sublyratus. Plastid localization and conversion to a farnesyl diphosphate synthase by mutagenesis. Chem Pharm Bull, 49 (2): 197-202. [PMID:11217109]
35. Tang M, Odejinmi SI, Allette YM, Vankayalapati H, Lai K. (2011) Identification of novel small molecule inhibitors of 4-diphosphocytidyl-2-C-methyl-D-erythritol (CDP-ME) kinase of Gram-negative bacteria. Bioorg Med Chem, 19 (19): 5886-95. [PMID:21903402]
36. Voynova NE, Rios SE, Miziorko HM. (2004) Staphylococcus aureus mevalonate kinase: isolation and characterization of an enzyme of the isoprenoid biosynthetic pathway. J Bacteriol, 186 (1): 61-7. [PMID:14679225]
37. Wakazono A, Fukao T, Yamaguchi S, Hori T, Orii T, Lambert M, Mitchell GA, Lee GW, Hashimoto T. (1995) Molecular, biochemical, and clinical characterization of mitochondrial acetoacetyl-coenzyme A thiolase deficiency in two further patients. Hum Mutat, 5 (1): 34-42. [PMID:7728148]
38. Waterham HR. (2002) Inherited disorders of cholesterol biosynthesis. Clin Genet, 61 (6): 393-403. [PMID:12121345]
39. Waterham HR. (2006) Defects of cholesterol biosynthesis. FEBS Lett, 580 (23): 5442-9. [PMID:16876788]
40. Weingärtner O, Lütjohann D, Böhm M, Laufs U. (2010) Relationship between cholesterol synthesis and intestinal absorption is associated with cardiovascular risk. Atherosclerosis, 210 (2): 362-5. [PMID:20116793]
41. Wiemer AJ, Tong H, Swanson KM, Hohl RJ. (2007) Digeranyl bisphosphonate inhibits geranylgeranyl pyrophosphate synthase. Biochem Biophys Res Commun, 353 (4): 921-5. [PMID:17208200]
42. Zhang C, Liu L, Xu H, Wei Z, Wang Y, Lin Y, Gong W. (2007) Crystal structures of human IPP isomerase: new insights into the catalytic mechanism. J Mol Biol, 366 (5): 1437-46. [PMID:17137593]
43. Zhang Y, Cao R, Yin F, Hudock MP, Guo RT, Krysiak K, Mukherjee S, Gao YG, Robinson H, Song Y et al.. (2009) Lipophilic bisphosphonates as dual farnesyl/geranylgeranyl diphosphate synthase inhibitors: an X-ray and NMR investigation. J Am Chem Soc, 131 (14): 5153-62. [PMID:19309137]
44. Zheng W, Sun F, Bartlam M, Li X, Li R, Rao Z. (2007) The crystal structure of human isopentenyl diphosphate isomerase at 1.7 A resolution reveals its catalytic mechanism in isoprenoid biosynthesis. J Mol Biol, 366 (5): 1447-58. [PMID:17250851]





{kind=link}